How Common Are The Sexual Side Effect Of Propecia
My grandfather is currently taking finasteride for prostate reasons. Even though it is the 5mg pill, I couldn’t help but notice that sexual side effects such as ‘impotence’ and ‘abnormal ejaculation’ were listed under ‘Common’. Not uncommon, not rare, but common! That being said, how ‘common’ is common? Are side effects in general rare, but sexual dysfunction is the most persistent of the side effects? When you quote a 2% chance of being subjected to sexual side effects, of what number is that 2% drawn from? Are sexual side effects rarer in
Propecia because of its smaller (1mg) dose?
Most of the studies on 1-2% sexual dysfunction is derived from the Prosar (finasteride 5mg dose). Taking a smaller dose should give you less side effect but the reported side effect is still around 1-2%.
This is taken directly from the MERCK website Propecia insert:
“In three controlled clinical trials for PROPECIA of 12-month duration, 1.4% of patients taking PROPECIA (n=945) were discontinued due to adverse experiences that were considered to be possibly, probably or definitely drug-related (1.6% for placebo; n=934). Clinical adverse experiences that were reported as possibly, probably or definitely drug-related in ?1% of patients treated with PROPECIA or placebo are presented in Table 1.”
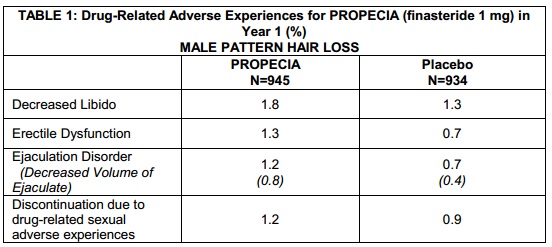
So per Merck the ‘drug-related adverse experiences’ causing patient trial discontinuation were marginally more common with *the placebo* than with finasteride, 1.6% vs 1.4%…?
Please note that regulatory definitions of frequency of adverse events (“side effects”) in clinical trials is somewhat different from what people think of these words colloquially.
For purposes of drug labeling, the frequency of occurrence of adverse events in a population of study patients is classified as:
Very common > 10% (of patients)
Common: > 1 to 10%
Uncommon: > 0.1 to 1%
Rare: > 0.01 to 0.1%
In addition, frquency of adverse event in a label is not related to degree of “persistence”. An untoward change from study baseline reported by a study participant is clasified as an adverse event if it is reported for ANY length of time (even 1 hour). The frequency in a label wil nopot distinhuish between short-yerm or longer events (classified the same). One needs to look at the study pblication or tyhe submiussion package (freely available on the FDA web site) for more detailed safety analyses, which typically contains info about duration of adverse events iof they do not “resolve” (ie all events are catgeorized as ongoing or resolved by study completion)
Sorry about the above spellings – there is no way to edit once submitted!