Drugs and processes that are developed to treat human conditions or diseases require that certain processes get followed to determine what I have stated over and over again — the safety and effectiveness of the drug or process. The research goes through many stages, and when the process reaches the clinical stages, the process gets very complex, expensive, and time consuming. So to elaborate upon what the FDA requires, I researched ‘what happens in a clinical trial’ to help my readers and patients understand what goes on, why it happens, and what is involved that takes so much time and money. I hope that the following piece (gleamed from an FDA publication — Inside Clinical Trials) sheds light on the subject. The question asks, “Why does the FDA slow down the process?” The answer below shows how the safety of the public is secured, defines the risks and benefits of the process/drug under study.
Every clinical trial is carefully designed to answer certain research questions. A trial plan called a protocol, maps out what study procedures will be done, by whom, and why. Products are often tested to see how they compare to standard treatments or to no treatment. The FDA often provides extensive technical assistance to researchers conducting clinical trials, helping them design better trials that can characterize effects of a new product more efficiently, while reducing risks to those participating in the trials.
The clinical trial team includes doctors and nurses, as well as other health care professionals. This team checks the health of the participant at the beginning of the trial and assesses whether that person is eligible to participate. Those found to be eligible–and who agree to participate–are given specific instructions, and then monitored and carefully assessed during the trial and after it is completed.
Done at hospitals and research centers around the country, clinical trials are conducted in phases. Phase 1 trials try to determine dosing, document how a drug is metabolized and excreted, and identify acute side effects. Usually, a small number of healthy volunteers (between 20 and 80) are used in Phase 1 trials.
Phase 2 trials include more participants (about 100-300) who have the disease or condition that the product potentially could treat. In Phase 2 trials, researchers seek to gather further safety data and preliminary evidence of the drug’s beneficial effects (efficacy), and they develop and refine research methods for future trials with this drug. If the Phase 2 trials indicate that the drug may be effective–and the risks are considered acceptable, given the observed efficacy and the severity of the disease–the drug moves to Phase 3.
In Phase 3 trials, the drug is studied in a larger number of people with the disease (approximately 1,000-3,000). This phase further tests the product’s effectiveness, monitors side effects and, in some cases, compares the product’s effects to a standard treatment, if one is already available. As more and more participants are tested over longer periods of time, the less common side effects are more likely to be revealed.
Sometimes, Phase 4 trials are conducted after a product is already approved and on the market to find out more about the treatment’s long-term risks, benefits, and optimal use, or to test the product in different populations of people, such as children.
Phase 2 and Phase 3 clinical trials generally involve a “control” standard. In many studies, one group of volunteers will be given an experimental or “test” drug or treatment, while the control group is given either a standard treatment for the illness or an inactive pill, liquid, or powder that has no treatment value (placebo). This control group provides a basis for comparison for assessing effects of the test treatment. In some studies, the control group will receive a placebo instead of an active drug or treatment. In other cases, it is considered unethical to use placebos, particularly if an effective treatment is available. Withholding treatment (even for a short time) would subject research participants to unreasonable risks.
The treatment each trial participant receives is often decided by a process called randomization. This process can be compared to a coin toss that is done by computer. During clinical trials, no one likely knows which therapy is better, and randomization assures that treatment selection will be free of any preference a physician may have. Randomization increases the likelihood that the groups of people receiving the test drug or control are comparable at the start of the trial, enabling comparisons in health status between groups of patients who participated in the trial.
In conjunction with randomization, a feature known as blinding helps ensure that bias doesn’t distort the conduct of a trial or the interpretation of its results. Single-blinding means the participant does not know whether he or she is receiving the experimental drug, an established treatment for that disease, or a placebo. In a single-blinded trial, the research team does know what the participant is receiving.
A double-blinded trial means that neither the participant nor the research team knows during the trial which participants receive the experimental drug. The patient will usually find out what he or she received at a pre-specified time in the trial.
 How did 16th Century playwright William Shakespeare lose his hair? I’m sure you find yourself pondering that question every waking day of your life. Well, perhaps not. At any rate, I stumbled upon this year and a half old article that has an interesting take on why Shakespeare possibly lost his hair. The title of the article gives away the punchline, but enjoy…
How did 16th Century playwright William Shakespeare lose his hair? I’m sure you find yourself pondering that question every waking day of your life. Well, perhaps not. At any rate, I stumbled upon this year and a half old article that has an interesting take on why Shakespeare possibly lost his hair. The title of the article gives away the punchline, but enjoy…
 The need to have luxurious hair is a quest that many people have and that is why the TV ads fill the airwaves with shampoos and conditioners, potions and lotions that are focused on people who do not like the texture of their hair or the dryness of it (or in your case the slimy nature of it). Hair does change texture with time and stress. It also changes if we expose it to the sun (drying it), chlorine from a swimming pool or other things. As we get older, hair clearly changes its character and its thickness and these two are absolutely related. I looked up Adipex and found references to a drug called Phentermine which is sold without a prescription. In the look-up, I also saw a link to this drug and hair loss with the Microsoft Diet on Google, and I got excited that Microsoft has found some magical cure to weight loss which I could tell my readership. Now is that getting us somewhere or isn’t it? I wonder if we can download a cure for hair loss from Microsoft if we buy into their diet (see
The need to have luxurious hair is a quest that many people have and that is why the TV ads fill the airwaves with shampoos and conditioners, potions and lotions that are focused on people who do not like the texture of their hair or the dryness of it (or in your case the slimy nature of it). Hair does change texture with time and stress. It also changes if we expose it to the sun (drying it), chlorine from a swimming pool or other things. As we get older, hair clearly changes its character and its thickness and these two are absolutely related. I looked up Adipex and found references to a drug called Phentermine which is sold without a prescription. In the look-up, I also saw a link to this drug and hair loss with the Microsoft Diet on Google, and I got excited that Microsoft has found some magical cure to weight loss which I could tell my readership. Now is that getting us somewhere or isn’t it? I wonder if we can download a cure for hair loss from Microsoft if we buy into their diet (see 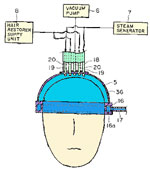 That is quite funny! Thanks for passing the link to me.
That is quite funny! Thanks for passing the link to me. I won’t comment on the doctor, but I will say that after 4 1/2 months you will not see the results of your surgery. The first and final element of what Dr. X did for you will be in the results that you get as compared with your expectations at about the 8th month after surgery.
I won’t comment on the doctor, but I will say that after 4 1/2 months you will not see the results of your surgery. The first and final element of what Dr. X did for you will be in the results that you get as compared with your expectations at about the 8th month after surgery.